Mélanie Hinterlang, Mona Assefi, Pauline Glasman, Johanne Silvain, Delphine Brugier, Marie Antignac, Fanny Charbonnier-Beaupel et Carole MetzComment citer cet article: Hinterlang M, Assefi M, Glasman P, Silvain J, Brugier D, Antignac M, et al. Sécurisation du circuit du médicament expérimental dans les services investigateurs en cas de dispensation non nominative par la méthode AMDEC. Can J Hosp Pharm. 2025;78(4):e3812. doi: 10.4212/cjhp.3812
RÉSUMÉ
Contexte
Les études cliniques en soins critiques nécessitent parfois des délais d’inclusion et d’administration très courts, pouvant avoir lieu à toute heure. Pour permettre d’optimiser la prise en charge des patients, le médicament expérimental peut être mis à disposition dans le service investigateur. Une analyse de risques de ce circuit a été réalisée afin de le sécuriser.
Objectif
Déterminer des axes de sécurisation du circuit de dispensation non nominative des médicaments expérimentaux pour les essais cliniques.
Méthodologie
À la suite d’une enquête préliminaire, 3 services pilotes ont été sélectionnés : la réanimation chirurgicale, la salle de surveillance post-interventionnelle (SSPI) et la cardiologie. La méthode d’analyse de risques AMDEC « Analyse des modes de défaillances, de leurs effets et de leur criticité » a été appliquée.
Résultats
Au total, 281 risques ont été identifiés. La majorité était acceptable, soit, 123 (44 %), 110 (39 %), 147 (52 %), ou tolérable, soit 139 (49 %), 148 (53 %) et 130 (46 %), en réanimation, SSPI et cardiologie respectivement. Les risques inacceptables étaient au nombre de 19 (7 %), 23 (8 %) et 4 (1 %) en réanimation, SSPI et cardiologie respectivement. Le processus identifié comme le plus critique pour les 3 services est la communication. Après priorisation des risques, 17 actions ont été proposées.
Conclusion
Cette étude a permis d’identifier des pistes d’intervention pour permettre la maîtrise du circuit de dispensation non nominative. Une fois les actions mises en place, une baisse de la criticité avec l’ensemble des risques acceptables ou tolérables est attendue. A long terme, ce projet a pour objectif d’améliorer la prise en charge des patients inclus dans les essais cliniques de soins d’urgence et de favoriser la recherche dans les services concernés.
Mots-clés: AMDEC, risque, sécurisation, médicament expérimental, soins critiques
ABSTRACT
Background
Clinical studies in critical care sometimes require very short time frames for study inclusion and drug administration, which may occur at any time. To optimize patient management, experimental drugs can be made directly available within the study unit.
Objective
To determine key areas of focus for controlling the non–patient-specific drug dispensing process for experimental drugs used in clinical trials.
Methods
After a preliminary survey, 3 pilot units were selected: the surgical intensive care unit, the post-intervention surveillance unit (PISU), and the cardiology unit. The failure modes, effects, and criticality analysis (FMECA) risk assessment method was applied.
Results
A total of 281 risks were identified. The majority were “acceptable” — 123 (44%), 110 (39%), and 147 (52%) — or “tolerable” — 139 (49%), 148 (53%), and 130 (46%) — in surgical intensive care, the PISU, and cardiology, respectively. The number of “unacceptable” risks was 19 (7%), 23 (8%), and 4 (1%) in the 3 units, respectively. Communication was identified as the most critical process across all 3 units. Following risk prioritization, 17 corrective measures were proposed.
Conclusions
This study helped identify potential areas for intervention to control the non–patient-specific drug dispensing process. Once the proposed actions are implemented, a reduction in overall risk criticality is expected, with all remaining risks falling within acceptable or tolerable levels. In the long term, this project aims to improve the management of patients enrolled in critical care clinical trials and promote research within the units involved.
Keywords: failure modes, effects, and criticality analysis (FMECA), risk, safety, experimental drug, critical care
Dans une étude clinique, chacune des étapes du circuit du médicament expérimental constitue un processus spécifique, qui le différencie de celui des soins courants, en raison du référentiel particulier auquel il est soumis : bonne pratique clinique1, European regulation2 en Europe, et chaque protocole spécifique d’étude clinique. Les modalités particulières de réception, de stockage, de conditionnement, de dispensation et de gestion des retours nécessitent une vigilance accrue à chacune de ces étapes, en particulier lorsqu’une gestion de l’insu s’impose.
En France, selon l’article L5126-1 du Code de la Santé Publique (CSP)3, la gestion des produits expérimentaux est une mission obligatoire des pharmacies à usage intérieur. Ces dernières doivent assurer la gestion, l’approvisionnement, la préparation, le contrôle, la détention, l’évaluation et la dispensation des médicaments, y compris des médicaments expérimentaux, tout en garantissant leur qualité.
Les études cliniques constituent une étape indispensable dans le développement et la vie d’un médicament. Ainsi, la recherche clinique est présente dans les centres hospitaliers universitaires, où elle occupe une place importante dans chaque service, y compris les services de soins d’urgence. Ces services de soins critiques doivent relever de nombreux défis dans le cadre de la recherche clinique, notamment la simplification excessive des interventions, la focalisation sur la mortalité comme critère principal, le caractère inflexible des protocoles, ainsi que les ressources et le temps nécessaires à la mise en œuvre et à la collecte des données4,5. Par ailleurs, leurs contraintes spécifiques nécessitent, pour certains essais cliniques, une accessibilité rapide et continue au médicament expérimental, 24 heures sur 24 et 7 jours sur 7. Dans ce contexte, une solution possible est d’utiliser un circuit de dispensation non nominative des médicaments expérimentaux, qui seront alors stockés directement dans le service investigateur. Dans ces cas-là, une bonne gestion des médicaments expérimentaux au sein du centre investigateur est essentielle pour garantir la sécurité des patients, la qualité des données et l’attractivité du site.
Face à ces exigences, il est apparu nécessaire de déterminer des axes de sécurisation du circuit de dispensation non nominative des médicaments expérimentaux pour les essais où ce circuit est le plus adapté. Pour atteindre cet objectif, une analyse de risques par la méthode AMDEC (« Analyse des modes de défaillances, de leurs effets et de leur criticité ») a été réalisée. Cette analyse adopte une approche globale couvrant l’ensemble du circuit, de la réception à la pharmacie au retour du conditionnement, en passant par l’administration au patient. Une telle approche globale d’analyse de risques a priori est notamment recommandée par des autorités françaises telle que l’Haute Autorité de Santé (HAS)6.
Un état des lieux des pratiques des CHU français a été réalisé par notre équipe au moyen d’un sondage auprès de la Commission des Pharmaciens de Centres Hospitaliers Universitaires (CPCHU) durant l’été 2021. Cette commission regroupe les pharmaciens des CHU français; elle est structurée en différents groupes de travail dont un relatif aux essais cliniques. Ce sondage, sous la forme d’une enquête simple, anonyme et à compléter en ligne, interrogeait succinctement le jugement quantitatif et qualitatif porté au processus de mise à disposition des médicaments expérimentaux de chaque centre hospitalier. Notre centre n’y a pas participé.
Pour réaliser cette AMDEC, 3 services pilotes ont été sélectionnés : un service de réanimation chirurgicale, une SSPI (salle de surveillance post-interventionnelle) et un service de cardiologie. L’unité des essais cliniques de la pharmacie pilotait le projet. Lors de la cartographie, le nombre d’études cliniques avec des dispensations non nominatives en cours était de : 5 en réanimation, 4 en SSPI, et 6 en cardiologie. Le groupe de travail, multidisciplinaire, comprenait des médecins investigateurs, des attachés de recherche clinique, un chef de projet, une infirmière (IDE) et des pharmaciens.
La répartition dans chaque groupe était la suivante : 1 chef de projet et 2 attachés de recherche clinique en cardiologie, 1 médecin et un attaché de recherche clinique au SSPI et 1 médecin, 1 infirmière et un attaché de recherche clinique en réanimation. Deux pharmaciens participaient à tous les groupes et toutes les étapes de tous les processus ont été évaluées par chaque groupe.
L’analyse de risque a été réalisée en accord avec les recommandations des ICH Q9 (International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Q9 guidelines on quality risk management)7. La méthode AMDEC a été utilisée6.
Le système analysé couvrait les étapes allant de la réception des médicaments expérimentaux (ME) à la pharmacie jusqu’au retour des médicaments expérimentaux administrés dans les services investigateurs. Une cartographie des processus et des risques associés du circuit a été réalisée à la suite d’une immersion dans chacun des 3 services pilotes. L’objectif était d’obtenir une cartographie la plus exhaustive possible et adaptée aux spécificités des 3 services.
La cotation des risques a été effectuée en ligne grâce à des questionnaires remplis par les participants au projet dans chaque service (attachés de recherche clinique, investigateurs, infirmière, pharmaciens). Afin de garantir que les questionnaires soient remplis comme il faut, des explications écrites, orales et vidéo ont été fournies. L’échelle de cotation, élaborée lors d’une précédente analyse de risque de notre unité8 et dont les définitions des niveaux de gravité ont été précisées pour cette étude, définissait les niveaux de fréquence, détectabilité et gravité. Chaque participant utilisait cette échelle pour évaluer les risques (tableau 1). La gravité des risques prenait en compte 3 référentiels avec 5 niveaux pour chaque référentiel : le patient, l’étude et le service. Les niveaux concernant l’étude intégraient la différence entre la perte de qualités ponctuelle (niveau 2), les déviations mineures (niveau 3) au protocole, les déviations majeures (niveau 4) et la sortie du patient de l’étude (niveau 5). Les déviations mineures sont définies par des exigences pratiques ou processus qui ne sont pas susceptibles d’avoir une incidence sur les droits, la sécurité ou le bien-être des sujets ou sur la qualité et l’intégrité des données, contrairement aux déviations majeures. Les niveaux concernant les patients différenciaient le retard de prise en charge ponctuel sans effet secondaire (niveau 2), une iatrogénie évitable sans hospitalisation nécessaire (niveau 3), une hospitalisation ou une prolongation d’hospitalisation sans mise en jeu du pronostic vital (niveau 4) et enfin la mise en jeu du pronostic vital (niveau 5). Enfin les niveaux de gravité du service prenaient en compte le temps de désorganisation (< 1 h, entre 1 h et 24 h, > 24 h, suspension de l’activité). Chaque participant évaluait uniquement les processus le concernant.
TABLEAU 1 Échelle de cotation des risques : fréquence, détectabilité et gravité; interprétation de la criticité en fonction de son score
Pour chaque service et chaque risque, la criticité, cotée par chaque participant, a été calculée. La moyenne des criticités et son écart-type ont été calculés. La criticité retenue correspondait à la borne supérieure de l’écart-type. Les risques ont ensuite été priorisés : une criticité ≥ 50 sur 125 était considérée comme inacceptable et donc prioritaire (tableau 1). Ce seuil, utilisé pour l’AMDEC de notre unité, a été jugé adapté pour cette étude. Les résultats ont été discutés avec le groupe de travail de chaque service afin de valider les cotations, d’identifier les actions existantes et de définir les actions à mettre en place. Une nouvelle cotation résiduelle des risques prioritaires a ensuite été réalisée en intégrant les actions proposées selon la même méthodologie que la première évaluation.
Dix-neuf pharmacies de CHU français sur les 26 sollicitées ont répondu au questionnaire de retour d’expérience. Parmi elles, 11 (58 %) géraient plus de 300 études cliniques en cours au moment du sondage. Un seul centre ne pratiquait pas de dispensation non nominative de médicaments expérimentaux. Pour les 18 autres centres ayant recours à des études cliniques en dispensation non nominative, le nombre d’études concernées dépassait 10 pour 9 centres (50 %) et 50 pour 3 d’entre eux (17 %). La gestion du circuit expérimental au sein des services était systématiquement organisée en collaboration avec la pharmacie. Par ailleurs, les non-conformités liées à ce circuit étaient rapportées à la pharmacie. Les incidents identifiés par le plus de centres étaient : l’utilisation du traitement de soins courants à la place du traitement expérimental (3 centres), l’absence de suivi des températures de stockage (2 centres), les erreurs d’attribution de traitement (4 centres).
Pour l’ensemble des pharmacies interrogées, les principales motivations pour la mise en place d’une dispensation non nominative étaient le délai court entre l’inclusion et l’administration ainsi que la possibilité d’inclusions 24 heures sur 24. Les services impliqués dans ce mode de dispensation étaient variés, les plus fréquemment cités étant les réanimations, les services de maladies infectieuses, les services de cardiologie, les urgences et les blocs opératoires (notamment pour des études cliniques avec des dispositifs médicaux).
Dans 84 % des cas (16/19), les pharmacies réalisaient des dispensations durant la nuit et le week-end. Pour 87,5 % (14/16) d’entre elles, ces dispensations impliquaient le pharmacien de garde. Ces dispensations étaient nominatives et nécessitaient la formation et l’habilitation de l’ensemble des pharmaciens de garde du site. La fréquence de ces dispensations variait de 1 à 2 par semaine à 1 à 5 par an. Parmi les centres concernés, 30 % (4 centres) ont signalé des non-conformités.
Au total, 26 % (5/19) des centres considéraient que la dispensation non nominative de médicaments expérimentaux n’était pas un mode de dispensation justifié. Ils pointaient une absence de maîtrise de ce circuit ou estimaient qu’il était dégradé, avec d’autres solutions possibles existantes pour la nuit et le week-end (comme l’intervention du pharmacien de garde).
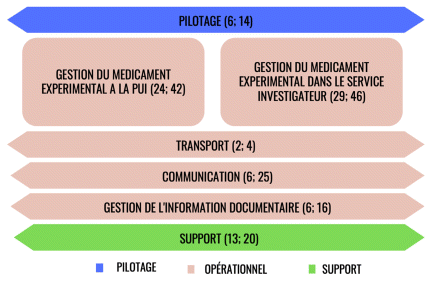
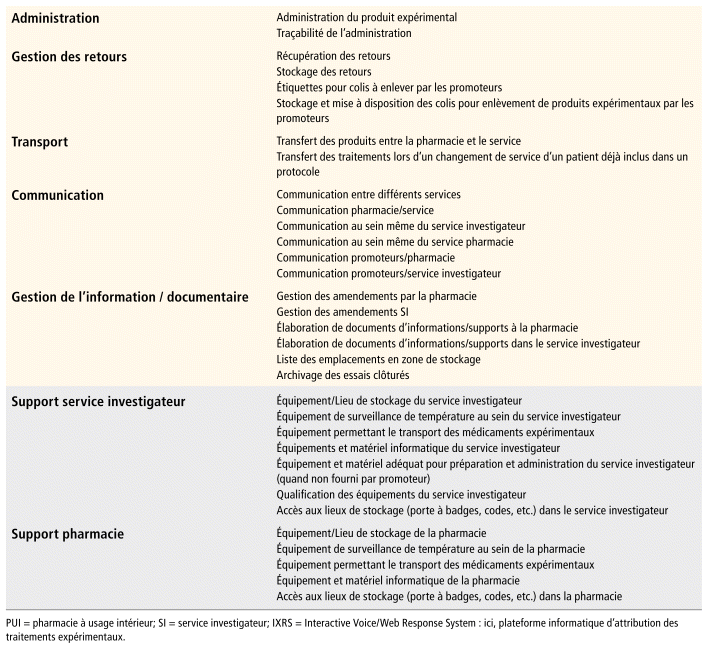
La cartographie du circuit de dispensation comprenait 7 processus principaux : pilotage (management et formation), gestion du médicament expérimental à la pharmacie, gestion du médicament expérimental dans le service investigateur, transport, communication, gestion de l’information documentaire et support (figure 1). Le processus de gestion du médicament expérimental dans le service investigateur englobait les étapes allant de la réception de la dotation à l’administration au patient, ainsi que le retour des traitements expérimentaux. Le processus de communication concernait les interactions au sein des services, entre les services et la pharmacie, et entre les services et les promoteurs et, le cas échéant, entre deux services investigateurs. Enfin, le processus support comprenait toutes les activités nécessaires pour la bonne réalisation des essais cliniques tel que les équipements ou la gestion des températures de stockage dans la pharmacie ou les services investigateurs. La gestion de l’information documentaire est le processus qui permet de documenter toutes les informations internes (p. ex., liste des emplacements des essais cliniques, documents d’informations/supports), mais aussi la gestion des informations externes (p. ex., gestion des amendements) (annexe 1).
|
| ||
|
FIGURE 1 Représentation des processus du circuit de dispensation non nominative des produits expérimentaux (PROCESSUS (n sous processus; n microprocessus)). PUI : pharmacie à usage intérieur. | ||
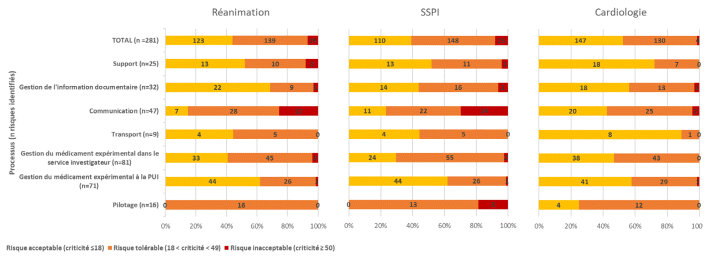
Ces 7 processus étaient déclinés en 84 sous-processus. Au total, 281 risques ont été identifiés dans la cartographie du circuit de dispensation non nominative des produits expérimentaux. Parmi les 281 risques identifiés, 19 (7 %), 23 (8 %) et 4 (1 %), sont ressortis avec une criticité inacceptable (≥ 50) à la suite de la cotation par les services de réanimation, SSPI et cardiologie respectivement (figure 2). Concernant les risques tolérables (criticité entre 18 et 49), ils étaient au nombre de 139 (49 %) en réanimation, 148 (53 %) en SSPI et 130 (46 %) en cardiologie. Enfin les risques acceptables (criticité < 18) étaient de 123 (44 %), 110 (39 %) et 147 (52 %) en réanimation, SPPI et cardiologie respectivement.
|
| ||
|
FIGURE 2 Répartition des risques par processus et criticité selon la cotation réalisée par les groupes de travail pour les services de réanimation chirurgicale, SSPI et cardiologie. PUI : pharmacie à usage intérieur; SSPI : salle de surveillance post-interventionnelle. | ||
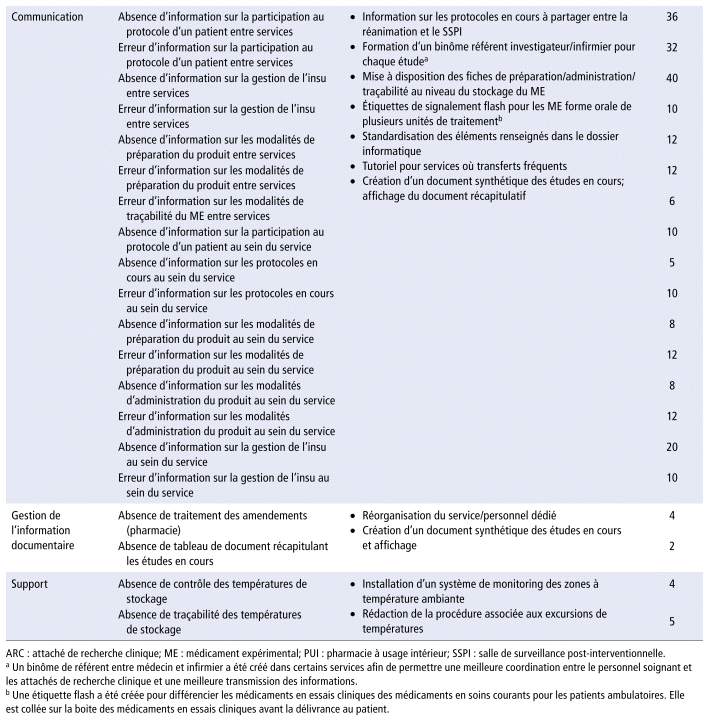
Les risques inacceptables concernaient principalement les processus opérationnels du circuit. Parmi eux, le processus de communication était systématiquement identifié comme ayant le plus de risques de criticité inacceptable dans les 3 services analysés. Certains risques inacceptables étaient communs aux services de réanimation et de SSPI, tandis que d’autres étaient spécifiques à la pharmacie, comme l’absence d’envoi de l’ordonnance préalablement remplie au service investigateur lors de la réception (processus de gestion du médicament expérimental à la pharmacie) et l’absence de traitement des amendements (processus de support).
Après l’identification des risques critiques, un plan d’actions a été élaboré pour intégrer des mesures préventives supplémentaires. Au total, 17 actions ont été définies. Certaines visent à prévenir plusieurs risques simultanément. Ces actions, détaillées dans le tableau 2, comprennent les risques inacceptables (criticité ≥ 50), les mesures proposées et la criticité résiduelle estimée après leur mise en œuvre. Certaines de ces actions sont déjà en cours d’application, tandis que d’autres restent à planifier.
TABLEAU 2 Risques inacceptables associés aux actions préventives proposées et leur cotation résiduelle pour chacun des processus

D’après l’enquête préliminaire, il apparaît clairement que les études cliniques menées en France adoptant un mode de dispensation non nominative sont loin d’être anecdotiques. Parmi les centres interrogés, 95 % (18/19) pratiquent ce mode de dispensation, dont 50 % pour plus de 10 études cliniques en cours. Ces données mettent en évidence l’importance de ce travail de sécurisation
Selon l’analyse de risques menée dans trois services pilotes (réanimation chirurgicale, SSPI et cardiologie), 281 risques ont été identifiés, parmi lesquels 19 (7 %), 23 (8 %) et 4 (1 %) ont été considérés comme inacceptables respectivement. À la suite de cette analyse, 17 actions ont été proposées pour réduire ces risques à un niveau tolérable ou acceptable.
Le processus opérationnel lié la communication est ressorti comme ayant le plus de risques inacceptables dans le circuit des médicaments expérimentaux en dispensation non nominative. Ce constat s’explique par les spécificités organisationnelles de chaque service. En SSPI, où les patients ne restent que quelques heures ou jours avant d’être transférés dans d’autres services, une communication efficace entre les équipes est déterminante. Cela explique un nombre légèrement plus élevé de risques inacceptables dans ce service par rapport à la réanimation ou à la cardiologie. En réanimation, les patients peuvent entrer dans le service en étant déjà inclus dans un essai clinique, mais sortent rarement du service en étant encore dans un protocole d’étude clinique nécessitant l’administration d’un médicament. En cardiologie, les patients sortent fréquemment avec leur traitement expérimental en ambulatoire. Cette diversité montre que, bien que les risques critiques liés à la communication soient communs aux trois services, leurs manifestations et leurs impacts diffèrent.
La méthode AMDEC a la capacité d’identifier et de prioriser les risques dans l’ensemble du processus et pas seulement de réagir aux événements indésirables déjà observés. Déjà utilisée dans le milieu hospitalier, notamment pour sécuriser le circuit du médicament9 ou du médicament expérimental10, cette méthode offre une approche globale applicable à l’ensemble du parcours des patients participant à des essais cliniques. Une revue de la littérature11 encourage d’ailleurs son utilisation pour cartographier et prioriser les risques à prévenir.
L’équipe du CHU de Strasbourg a appliqué cette méthode à la dispensation nominative des médicaments expérimentaux à la pharmacie de l’ouverture à la clôture de l’essai10. Parmi les défaillances identifiées, les problèmes de communication figuraient parmi les causes les plus fréquentes. Cela corrobore nos résultats, où la communication est également ressortie comme le processus présentant le plus de risques critiques. L’équipe de Strasbourg a proposé des solutions telles que la conception de documents d’informations. De manière similaire, les équipes de notre CHU ont élaboré des tutoriels et fiches récapitulatives sur la préparation, l’administration et la traçabilité des médicaments expérimentaux afin de faciliter et standardiser les transmissions d’information.
Bien que performante, l’AMDEC présente certaines limites. La subjectivité des cotations est une des limites. Les évaluations sont influencées par le vécu et les expériences des participants, introduisant un biais potentiel. Par ailleurs, la mise en place de cette méthode est longue et fastidieuse, mais elle favorise l’implication des acteurs concernés et les sensibilise aux risques. Dans cette étude, seule une infirmière a participé, et uniquement pour le service de réanimation. Bien que travailler avec trois services ait enrichi l’analyse, cela a complexifié la cartographie des processus, qui devait être adaptée à tous les services tout en restant généralisable. De plus, la composition de nos groupes de travail étant hétérogène, ce biais de recrutement a pu avoir une incidence sur les résultats.
Les risques identifiés qui présentaient une gravité > 4 pour l’un des trois référentiels (patient, étude, service) ont été isolés. Comme les autres risques identifiés, ils n’ont été priorisés que si leur criticité était supérieure à 50, mais ils feront l’objet d’une attention particulière dans le suivi au vu de leur gravité potentielle. Pour donner un exemple de ces risques, dans le sous processus opérationnel « administration », le risque « administration du mauvais produit expérimental » a été évalué avec une gravité > 4.
Les actions proposées, en cours de mise en place ou à planifier, nécessiteront une évaluation pour mesurer leur impact réel. Certaines entraineront des adaptations organisationnelles. Des temps de formation des équipes devront être aménagés pour les services où ce n’est pas déjà le cas. La mise en place d’un binôme investigateur/infirmière sera une nouveauté qui impliquera de définir des objectifs et de communiquer sur son rôle. Ces binômes pourraient améliorer la communication et réduire les risques, notamment en permettant un dialogue direct et spécifique autour des études cliniques. Lors des immersions dans les services, les témoignages d’IDE recueillis indiquaient tous un désir de comprendre a minima ce qu’ils préparent, administrent et dans quel but. Les formations pourront permettre, entre autres, de comprendre l’importance du recueil des données, de la précision attendue des études cliniques, mais aussi d’informer sur les études en cours dans les services. Le binôme permettra d’avoir un interlocuteur privilégié pour les questions pratiques plus précises et permettra d’améliorer la communication sur l’étude de façon plus ou moins importante en fonction de l’implication du binôme.
Les listes de contrôle, déjà utilisées avant la mise en place des études, devront intégrer des exigences spécifiques au circuit de mise à disposition. Elles permettront de vérifier que les barrières nécessaires sont instaurées et que la documentation est accessible à tous les acteurs.
L’analyse des risques a permis de recenser des pistes d’intervention pour sécuriser le circuit de dispensation non nominative. Bien qu’il soit réservé à des études nécessitant des inclusions et administrations de traitements en urgence, cette sécurisation pourrait encourager la recherche clinique dans les services de soins critiques.
En pratique, cette démarche aidera les services investigateurs à mettre en place ce type de circuit. Elle devra être complétée par une analyse des effets a posteriori des actions mises en place pour en évaluer l’efficacité sur la sécurisation du circuit du médicament expérimental.
1 Guideline for good clinical practice E6(R2). European Medicines Agency; 14 juin 2017 [consulté le 18 janvier 2024]. Disponible à : https://www.ema.europa.eu/en/ich-e6-r2-good-clinical-practice-scientific-guideline
2 Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC [European regulation]. 16 avr. 2014 [consulté le 20 janvier 2024]. Disponible à : https://eur-lex.europa.eu/eli/reg/2014/536/2022-12-05
3 Article L5126-1 – Code de la santé publique. Légifrance; 25 déc. 2022 [consulté le 12 décembre 2023]. Disponible à : https://www.legifrance.gouv.fr/codes/article_lc/LEGIARTI000045630266/
4 Granholm A, Alhazzani W, Derde LPG, Angus DC, Zampieri FG, Hammond NE, et al. Randomised clinical trials in critical care: past, present and future. Intensive Care Med. 2022;48(2):16478.
Crossref
5 Laffey JG, Kavanagh BP. Negative trials in critical care: why most research is probably wrong. Lancet Respir Med. 2018;6(9):65960.
Crossref
6 La sécurité des patients. Mettre en œuvre la gestion des risques associés aux soins en établissement de santé. Haute Autorité de Santé; mars 2012 [consulté le 14 septembre 2023]. Disponible à : https://www.has-sante.fr/jcms/c_1239410/fr/mettre-en-oeuvre-la-gestion-des-risques-associes-aux-soins-en-etablissement-de-sante
7 ICH guideline Q9 on quality risk management. European Medicines Agency; 26 juil. 2023 [consulté le 14 septembre 2023]. Disponible à : https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use-ich-guideline-q9-quality-risk-management-step-5-first-version_en.pdf
8 Sternjacob T. Mise en place d’une démarche de certification ISO 9001 au sein de l’unité de recherche clinique et innovation de la pharmacie à usage intérieur de l’hôpital universitaire de La Pitié-Salpêtrière (AP-HP. 2019). Thèse.
9 Abbassi A, Ben Cheikh Brahim A, Ouahchi Z. Failure mode and effect analysis applied to improve the medication management process in a pharmacy of a teaching hospital and a proposal for a simplified rating system. Eur J Hosp Pharm. 2023;30(e1):e5560.
Crossref
10 Hurtrel F, Beretz L, Renard V, Hutt A. Analyse des risques liés au circuit de gestion et de dispensation des produits en expérimentation clinique par «AMDEC». Risques qual milieu soins. 2012;9(1):22–30.
11 Demers É, Collin-Lévesque L, Boulé M, Lachapelle S, Nguyen C, Lebel D, et al. Analyse des modes de défaillance, de leurs effets et de leur criticité dans le circuit du médicament : revue de littérature. Can J Hosp Pharm. 2018;71(6):37684.
Adresse de correspondance : Mélanie Hinterlang, Unité REQPHARM, pharmacie à usage intérieur, Groupe Hospitalier Pitié-Salpêtrière, APHP, 47-83 Bd de l’Hôpital, 75013, Paris, France, Courriel: melanie.hinterlang@aphp.fr
Conflits d’intérêts: Aucune déclaration.
Financement: Aucun reçu.
Remerciements: Nous remercions toute l’équipe de l’unité REQPHARM qui a rendu ce travail possible ainsi que le service de réanimation chirurgicale du Pr Constantin, la SSPI du Pr Raux et le groupe Action Cœur des services de cardiologie pour leur accueil et leur participation active à cette analyse.
Soumis : 22 mars 2025
Accepté : 27 juin 2025
Publié : 10 décembre 2025

© 2025 Canadian Society of Healthcare-Systems Pharmacy | Société canadienne de pharmacie dans les réseaux de la santé
Canadian Journal of Hospital Pharmacy, VOLUME 78, NUMBER 4, 2025